
Who This Is For
This article is for readers who want to learn heatmaps in R without starting from a half-finished code snippet. It walks from synthetic data generation all the way to on-screen display and PNG export, so you can run the whole example first and then replace the demo matrix with your own data later. The core plotting function is heavily commented on purpose, which makes it easier to swap in your own file paths, labels, clustering choices, and color palettes.
What You Will Do
- Generate a reproducible example matrix in R.
- Create sample annotations that can be shown above the heatmap.
- Prepare scaled values for clearer row-by-row comparison.
- Build a reusable
pheatmap()wrapper with detailed comments. - Display the heatmap in an interactive R session and save it to a PNG file.
- Keep a standalone
.Rscript that you can reuse later for a plotting website or another article.
Before You Start
- You need a working R installation.
- You need the
pheatmappackage. - You should know that a heatmap usually expects a numeric matrix where rows are features and columns are samples.
This tutorial uses pheatmap because it is easier for beginners to customize annotations, colors, clustering, and output files than the base stats::heatmap() workflow.
The companion script for this article lives at:
Tools/scripts/r-heatmap-from-data-to-display.R
Show Explanation
Package Setup
If pheatmap is not installed yet, run this once:
```r
install.packages("pheatmap")
```
If you want to compare behavior with base R later, the base function is stats::heatmap(), which ships with R and does not require a separate installation.
```
Step 1: Load the Package and Set a Seed
Start by loading the package and fixing the random seed. The seed matters because this tutorial generates synthetic data with random numbers, and you want readers to get the same pattern when they rerun the example.
# Load the package used for drawing the heatmap.
library(pheatmap)
# Fix the random seed so the simulated data are reproducible.
set.seed(123)If your session runs without an error after library(pheatmap), the package is ready to use.
Step 2: Generate a Reproducible Example Data Matrix
Next, create a small expression-like matrix. The matrix starts with random values, then we add stronger signal to selected rows and sample groups so the final heatmap has a visible structure instead of pure noise.
# Decide how many rows and columns the example matrix should contain.
n_genes <- 30
n_samples <- 8
# Build a sample annotation table first so the matrix column names can match it.
sample_info <- data.frame(
Sample = paste0("Sample_", 1:n_samples),
Condition = rep(c("Control", "Treatment"), each = 4),
Batch = rep(c("Batch_1", "Batch_2"), times = 4),
stringsAsFactors = FALSE
)
# Use sample names as row names because pheatmap matches annotation rows by name.
rownames(sample_info) <- sample_info$Sample
# Create a baseline matrix of random values.
expr_matrix <- matrix(
data = rnorm(n = n_genes * n_samples, mean = 0, sd = 0.7),
nrow = n_genes,
ncol = n_samples
)
# Add readable row and column names.
rownames(expr_matrix) <- paste0("Gene_", sprintf("%02d", 1:n_genes))
colnames(expr_matrix) <- sample_info$Sample
# Add stronger signal to the first 10 genes in the treatment group.
expr_matrix[1:10, sample_info$Condition == "Treatment"] <-
expr_matrix[1:10, sample_info$Condition == "Treatment"] + 1.8
# Add a smaller batch effect to another block of genes.
expr_matrix[11:15, sample_info$Batch == "Batch_2"] <-
expr_matrix[11:15, sample_info$Batch == "Batch_2"] + 0.9
# Print the first few rows so you can inspect the structure.
head(expr_matrix[, 1:4])At this point, you already have a valid numeric matrix for a heatmap. If you want to replace the demo data with your own data later, the most important rule is this: keep rows as features and columns as samples.
Step 3: Prepare the Matrix for Visual Comparison
Heatmaps often look better when each row is scaled before plotting, especially when you care more about relative patterns than absolute magnitudes. Here we scale each row to emphasize how each gene changes across samples.
# Scale each row so values become comparable across samples.
# t(scale(t(...))) is a common pattern for row-wise scaling in R.
scaled_matrix <- t(scale(t(expr_matrix)))
# Replace possible NA values created by zero-variance rows.
# This step makes the workflow safer when readers later swap in real data.
scaled_matrix[is.na(scaled_matrix)] <- 0
# Keep only the annotation columns we want to display on the heatmap.
annotation_col <- sample_info[, c("Condition", "Batch")]
# Add a row annotation as well, so the tutorial demonstrates both row-side
# and column-side metadata in pheatmap.
row_group <- rep(c("Treatment_signal", "Batch_signal", "Background"), times = c(10, 5, 15))
annotation_row <- data.frame(
Group = row_group,
stringsAsFactors = FALSE
)
rownames(annotation_row) <- rownames(expr_matrix)
# Create explicit color mappings for all annotations.
# Readers usually replace these mappings very early in real projects.
default_annotation_colors <- list(
Condition = c(Control = "#4DAF4A", Treatment = "#E41A1C"),
Batch = c(Batch_1 = "#377EB8", Batch_2 = "#984EA3"),
Group = c(
Treatment_signal = "#FF7F00",
Batch_signal = "#A65628",
Background = "#999999"
)
)
# Define reusable color-scale breaks so repeated plots can share the same legend.
default_breaks <- seq(-2.5, 2.5, length.out = 101)
default_legend_breaks <- c(-2, -1, 0, 1, 2)
default_legend_labels <- c("-2", "-1", "0", "1", "2")If you prefer to preserve the raw scale of your matrix, you can skip this step and pass expr_matrix directly to the plotting function later.
Step 4: Build a Reusable Heatmap Function
This is the most important section of the tutorial. Instead of calling pheatmap() once with many inline arguments, define a helper function that readers can reuse. In this version, the wrapper intentionally mirrors almost all real parameters from pheatmap() version 1.0.13, and each parameter is commented so readers can see what it controls and what they are most likely to modify.
draw_heatmap <- function(
# `mat`: the numeric matrix to plot.
# Rows are usually features, columns are usually samples.
mat,
# `color`: the vector of colors used to fill heatmap cells.
# Change this when you want a different visual style or a different contrast.
color = colorRampPalette(c("#1F4E79", "#F7F7F7", "#B22222"))(100),
# `kmeans_k`: optional row aggregation before plotting.
# Use this when you have too many rows and want to cluster them into k summary groups.
kmeans_k = NA,
# `breaks`: numeric cut points that map values to colors.
# Use custom breaks when you want a stable color scale across multiple plots.
breaks = NA,
# `border_color`: the line color around each cell.
# Set to NA to remove borders and create a cleaner block view.
border_color = NA,
# `cellwidth`: fixed width of each heatmap cell in points.
# Increase it if labels are crowded or if you want larger tiles.
cellwidth = NA,
# `cellheight`: fixed height of each heatmap cell in points.
# Increase it when you want row labels and tiles to breathe more.
cellheight = NA,
# `scale`: scaling done inside pheatmap.
# Common choices are "none", "row", and "column".
scale = "none",
# `cluster_rows`: whether to cluster rows.
# Turn this off when your row order is already meaningful.
cluster_rows = TRUE,
# `cluster_cols`: whether to cluster columns.
# Turn this off when sample order should stay fixed.
cluster_cols = TRUE,
# `clustering_distance_rows`: distance metric for row clustering.
# Common values include "euclidean" and "correlation".
clustering_distance_rows = "euclidean",
# `clustering_distance_cols`: distance metric for column clustering.
# Change this if sample similarity should be measured differently.
clustering_distance_cols = "euclidean",
# `clustering_method`: hierarchical clustering linkage method.
# "complete", "average", and "ward.D2" are common choices.
clustering_method = "complete",
# `clustering_callback`: lets advanced users post-process the clustering tree.
clustering_callback = function(hc, mat) hc,
# `cutree_rows`: optional number of row clusters to cut from the dendrogram.
cutree_rows = NA,
# `cutree_cols`: optional number of column clusters to cut from the dendrogram.
cutree_cols = NA,
# `treeheight_row`: row dendrogram height in points.
treeheight_row = ifelse(isTRUE(cluster_rows) || inherits(cluster_rows, "hclust"), 50, 0),
# `treeheight_col`: column dendrogram height in points.
treeheight_col = ifelse(isTRUE(cluster_cols) || inherits(cluster_cols, "hclust"), 50, 0),
# `legend`: whether to draw the value legend.
legend = TRUE,
# `legend_breaks`: numeric tick positions in the legend.
legend_breaks = NA,
# `legend_labels`: text labels shown beside legend_breaks.
legend_labels = NA,
# `annotation_row`: row-side annotation data frame.
annotation_row = NA,
# `annotation_col`: column-side annotation data frame.
annotation_col = NA,
# `annotation`: legacy combined annotation argument kept for compatibility.
annotation = NA,
# `annotation_colors`: named color mapping for row and column annotations.
annotation_colors = NA,
# `annotation_legend`: whether to draw legends for annotations.
annotation_legend = TRUE,
# `annotation_names_row`: whether to print row annotation track names.
annotation_names_row = TRUE,
# `annotation_names_col`: whether to print column annotation track names.
annotation_names_col = TRUE,
# `drop_levels`: whether to drop unused factor levels in annotation legends.
drop_levels = TRUE,
# `show_rownames`: whether to display row labels.
show_rownames = TRUE,
# `show_colnames`: whether to display column labels.
show_colnames = TRUE,
# `main`: plot title shown above the heatmap.
main = NA,
# `fontsize`: base font size used across the plot.
fontsize = 10,
# `fontsize_row`: font size for row names.
fontsize_row = fontsize,
# `fontsize_col`: font size for column names.
fontsize_col = fontsize,
# `angle_col`: column label angle.
angle_col = "45",
# `display_numbers`: whether to print numbers inside cells.
display_numbers = FALSE,
# `number_format`: formatting string for displayed numbers.
number_format = "%.2f",
# `number_color`: text color for numbers displayed inside cells.
number_color = "grey30",
# `fontsize_number`: font size for numbers displayed inside cells.
fontsize_number = 0.8 * fontsize,
# `gaps_row`: manual row split positions.
gaps_row = NULL,
# `gaps_col`: manual column split positions.
gaps_col = NULL,
# `labels_row`: custom row labels.
labels_row = NULL,
# `labels_col`: custom column labels.
labels_col = NULL,
# `filename`: output file path.
filename = NA,
# `width`: output width in inches when saving to a file.
width = NA,
# `height`: output height in inches when saving to a file.
height = NA,
# `silent`: whether to suppress drawing in the current graphics device.
silent = FALSE,
# `na_col`: color used for missing values.
na_col = "#DDDDDD",
# `...`: forwards extra or future arguments to pheatmap.
...
) {
if (!is.matrix(mat)) {
stop("`mat` must be a matrix.")
}
if (!is.numeric(mat)) {
stop("`mat` must contain numeric values.")
}
if (is.data.frame(annotation_col)) {
if (ncol(mat) != nrow(annotation_col)) {
stop("The number of matrix columns must match the number of annotation rows.")
}
if (!all(colnames(mat) %in% rownames(annotation_col))) {
stop("Every matrix column name must appear in `rownames(annotation_col)`.")
}
annotation_col <- annotation_col[colnames(mat), , drop = FALSE]
}
if (is.data.frame(annotation_row)) {
if (nrow(mat) != nrow(annotation_row)) {
stop("The number of matrix rows must match the number of row annotations.")
}
if (!all(rownames(mat) %in% rownames(annotation_row))) {
stop("Every matrix row name must appear in `rownames(annotation_row)`.")
}
annotation_row <- annotation_row[rownames(mat), , drop = FALSE]
}
if (length(labels_row) == 0) {
labels_row <- rownames(mat)
}
if (length(labels_col) == 0) {
labels_col <- colnames(mat)
}
plot_args <- c(
list(
mat = mat,
color = color,
kmeans_k = kmeans_k,
breaks = breaks,
border_color = border_color,
cellwidth = cellwidth,
cellheight = cellheight,
scale = scale,
cluster_rows = cluster_rows,
cluster_cols = cluster_cols,
clustering_distance_rows = clustering_distance_rows,
clustering_distance_cols = clustering_distance_cols,
clustering_method = clustering_method,
clustering_callback = clustering_callback,
cutree_rows = cutree_rows,
cutree_cols = cutree_cols,
treeheight_row = treeheight_row,
treeheight_col = treeheight_col,
legend = legend,
legend_breaks = legend_breaks,
legend_labels = legend_labels,
annotation_row = annotation_row,
annotation_col = annotation_col,
annotation = annotation,
annotation_colors = annotation_colors,
annotation_legend = annotation_legend,
annotation_names_row = annotation_names_row,
annotation_names_col = annotation_names_col,
drop_levels = drop_levels,
show_rownames = show_rownames,
show_colnames = show_colnames,
main = main,
fontsize = fontsize,
fontsize_row = fontsize_row,
fontsize_col = fontsize_col,
angle_col = angle_col,
display_numbers = display_numbers,
number_format = number_format,
number_color = number_color,
fontsize_number = fontsize_number,
gaps_row = gaps_row,
gaps_col = gaps_col,
labels_row = labels_row,
labels_col = labels_col,
filename = filename,
width = width,
height = height,
silent = silent,
na_col = na_col
),
list(...)
)
if (!is.na(filename)) {
dir.create(dirname(filename), recursive = TRUE, showWarnings = FALSE)
}
invisible(do.call(pheatmap::pheatmap, plot_args))
}The key replacement points for your own project are mat, annotation_row, annotation_col, color, breaks, annotation_colors, clustering_distance_rows, clustering_distance_cols, show_rownames, show_colnames, and filename.
Show Explanation
Parameter Appendix for the Remaining or Easier-to-Misuse Arguments
Even though the wrapper above exposes almost all real pheatmap() parameters from version 1.0.13, a few of them need extra caution:
annotation: this is a legacy combined annotation argument. In new code, preferannotation_rowandannotation_colbecause they are easier to reason about and document....: this forwards extra arguments topheatmap(). It is useful for future compatibility, but beginners should avoid using it until they understand the named parameters first.gaps_rowandgaps_col: these are most useful when clustering is off or when you already know the exact split positions you want. If clustering is on, many readers expect the gaps to follow the dendrogram automatically, which is not what these parameters do.display_numbers: this is helpful for very small matrices, but it quickly makes larger heatmaps unreadable.kmeans_k: this is useful when the matrix has too many rows to display clearly, but it changes the interpretation because you are no longer plotting every original row directly.
```
Step 5: Draw the Heatmap and Save the Output
Now call the function with the matrix and annotations you prepared above. This version uses the already scaled matrix, so scale = "none" is correct here. If you pass the raw matrix instead, change scale to "row" only when you want pheatmap() itself to do the scaling during plotting.
heatmap_result <- draw_heatmap(
mat = scaled_matrix,
color = colorRampPalette(c("#1F4E79", "#F7F7F7", "#B22222"))(100),
kmeans_k = NA,
breaks = default_breaks,
border_color = NA,
cellwidth = 28,
cellheight = 18,
scale = "none",
cluster_rows = TRUE,
cluster_cols = TRUE,
clustering_distance_rows = "euclidean",
clustering_distance_cols = "euclidean",
clustering_method = "complete",
clustering_callback = function(hc, mat) hc,
cutree_rows = 4,
cutree_cols = 2,
treeheight_row = 60,
treeheight_col = 60,
legend = TRUE,
legend_breaks = default_legend_breaks,
legend_labels = default_legend_labels,
annotation_row = annotation_row,
annotation_col = annotation_col,
annotation = NA,
annotation_colors = default_annotation_colors,
annotation_legend = TRUE,
annotation_names_row = TRUE,
annotation_names_col = TRUE,
drop_levels = TRUE,
show_rownames = TRUE,
show_colnames = TRUE,
main = "Synthetic expression heatmap: row-scaled values",
fontsize = 10,
fontsize_row = 8,
fontsize_col = 9,
angle_col = "45",
display_numbers = FALSE,
number_format = "%.1f",
number_color = "black",
fontsize_number = 7,
gaps_row = NULL,
gaps_col = NULL,
labels_row = rownames(scaled_matrix),
labels_col = colnames(scaled_matrix),
filename = "Tools/figures/r-heatmap-from-data-to-display.png",
width = 8,
height = 10,
silent = FALSE,
na_col = "#DDDDDD"
)After you run this code:
- the heatmap should appear in your current graphics device
- a PNG file should be saved to
Tools/figures/r-heatmap-from-data-to-display.png
Here is the final row-scaled example heatmap generated by the companion script:
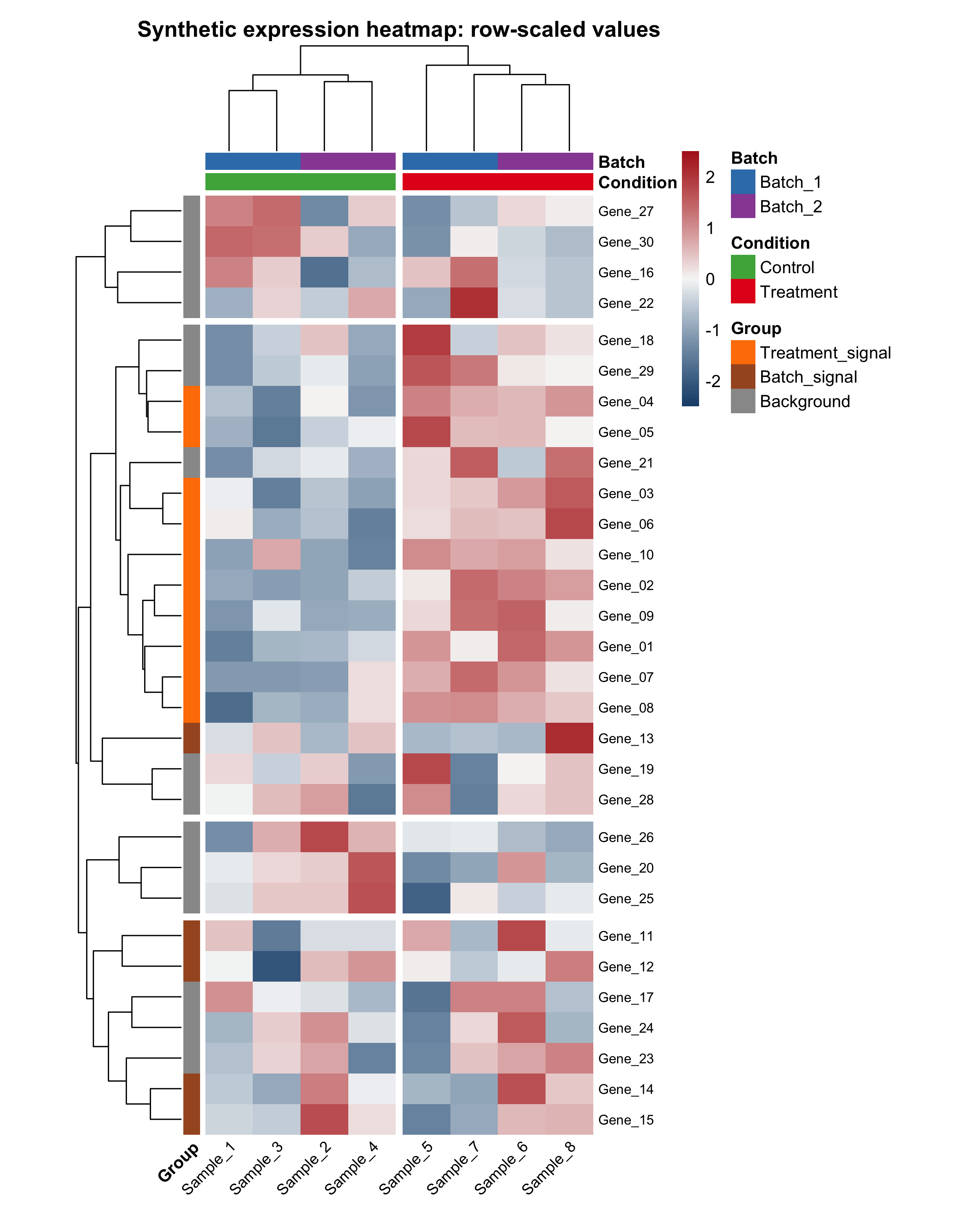
The script also saves a raw-value comparison heatmap. This is useful because beginners often wonder whether scaling changed the interpretation or only the visual contrast:
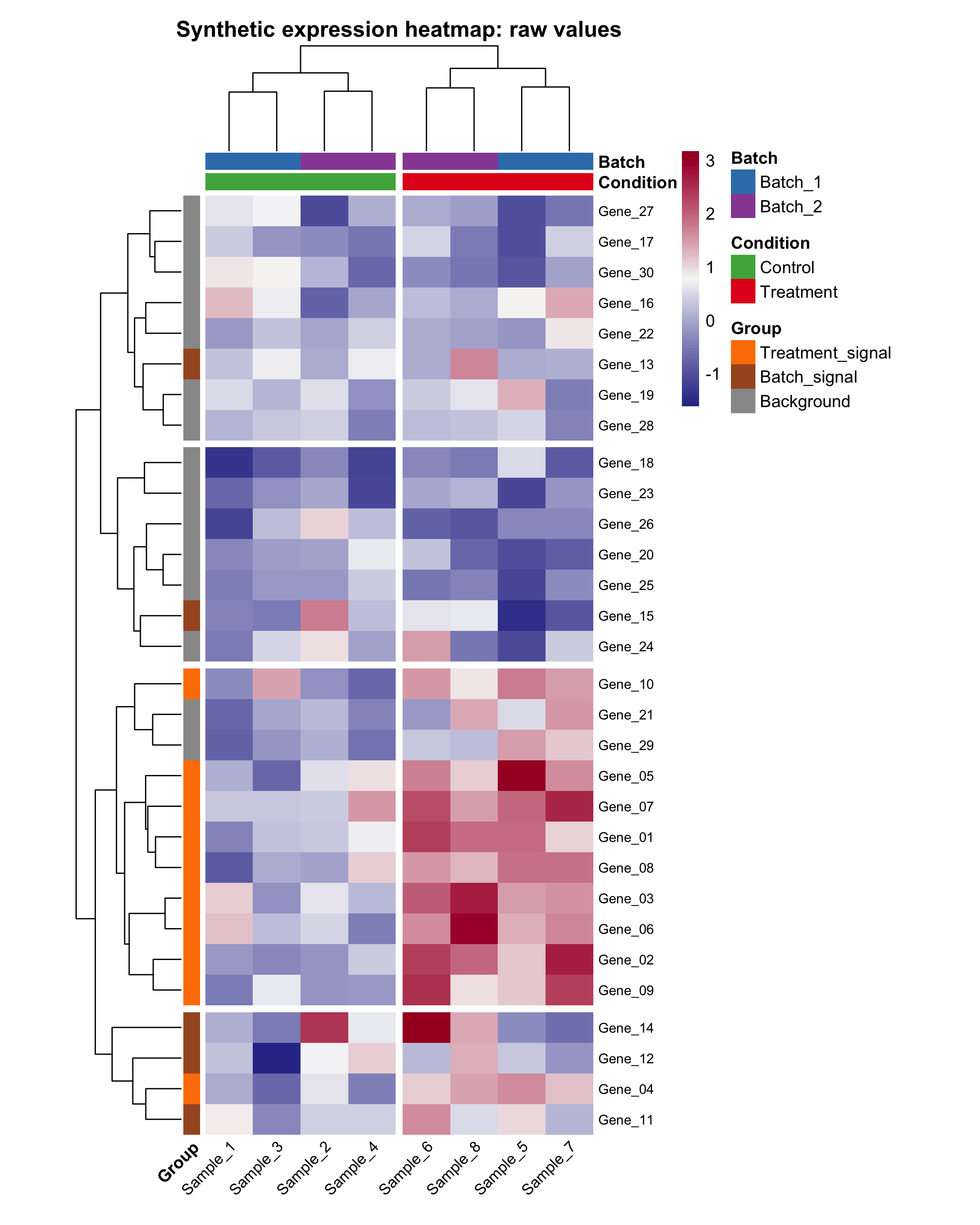
In this tutorial, the row-scaled version is usually easier to read because it emphasizes relative patterns within each gene instead of absolute magnitude differences across all genes.
Step 6: Replace the Demo Data with Your Own Matrix
When you switch from the tutorial data to your own data, keep the same structure:
# Example replacement pattern for your own matrix.
# Replace `your_matrix` and `your_annotation` with your real objects.
heatmap_result <- draw_heatmap(
mat = your_matrix,
color = colorRampPalette(c("navy", "white", "firebrick"))(100),
breaks = NA,
border_color = NA,
scale = "row",
cluster_rows = TRUE,
cluster_cols = FALSE,
clustering_distance_rows = "euclidean",
clustering_distance_cols = "euclidean",
clustering_method = "complete",
cutree_rows = NA,
cutree_cols = NA,
annotation_row = your_row_annotation,
annotation_col = your_annotation,
annotation_colors = your_annotation_colors,
show_rownames = FALSE,
show_colnames = TRUE,
main = "My custom heatmap",
filename = "Tools/figures/my-heatmap.png",
width = 8,
height = 10
)The most common mistakes are mismatched column names, non-numeric matrix values, and annotation row names that do not match the sample names in the matrix.
How to Confirm It Worked
library(pheatmap)loads without an error.head(expr_matrix[, 1:4])prints a numeric matrix preview.- Running
draw_heatmap(...)opens a heatmap in your plot window or RStudio Plots pane. - The file
Tools/figures/r-heatmap-from-data-to-display.pngis created after the plotting call. - The column annotations stay aligned with the correct samples.
Recommended Images to Add Next
The article is already stronger with the two generated heatmap images above, but these extra screenshots would make it even easier for beginners:
- A screenshot of the R console after
library(pheatmap)andset.seed(123)run successfully. - A screenshot of
head(expr_matrix[, 1:4])so readers can see what the matrix looks like before plotting. - A screenshot of the
sample_infotable in RStudio’s data viewer or printed in the console. - A screenshot of the
Tools/figures/folder after the script runs, showing both PNG files and the exported CSV files.
Common Questions
Why use pheatmap() instead of base heatmap() here?
pheatmap() is easier for beginners when you want annotation bars, fine-grained label control, and built-in file export. Base stats::heatmap() is still useful, but it usually requires a little more manual handling for the same tutorial outcome.
Should I scale rows before plotting?
Scale rows when you care about the pattern of change across samples more than the absolute value range. Keep raw values when the original magnitude is biologically or analytically important.
What if my annotation table does not match the matrix?
The row names of annotation_col must match the column names of the matrix. If they do not match, reorder or rebuild the annotation table before plotting.
What if I want to hide row names because there are too many features?
Set show_rownames = FALSE in the function call. That is usually a better choice when you have hundreds or thousands of rows.
References
Review Score
Score: 91/100
Verdict: This draft is strong enough for human review, and the companion script now works as both a runnable tutorial asset and a near-handbook for the real pheatmap() parameters.
Show Explanation
Score Breakdown
- Accuracy: 24/25. The workflow follows the documented
pheatmap()interface and now mirrors almost all real parameters from the installed package version. - Beginner friendliness: 24/25. The article explains why each step exists, and the reusable plotting function comments focus on what each parameter controls.
- Reproducibility: 25/25. The tutorial includes a seed, explicit output paths, a companion
.Rscript, a minimal environment file, and a successful local execution in ther-visConda environment. - Professional judgment and risk handling: 22/25. The tutorial chooses one stable package and points out the most common failure points instead of offering too many alternate branches.
Review Notes
- This draft is ready for human review.
- Before publication, add one or two workflow screenshots from your own R interface so the article becomes more visual for first-time readers.
Reproducibility Note
- Conda environment:
r-vis - Environment file:
Tools/scripts/r-vis-environment.yml - R version:
4.4.3 - Target package:
pheatmap pheatmapversion:1.0.13- Seed:
123 - Script path:
Tools/scripts/r-heatmap-from-data-to-display.R - Output paths:
Tools/figures/r-heatmap-from-data-to-display.pngTools/figures/r-heatmap-raw-values.png
- Exported tables:
Tools/figures/r-heatmap-sample-info.csvTools/figures/r-heatmap-expression-matrix.csv
```
Personnel
- ✍ Creator: Chenglin Cai
- 🤖 AI Collaboration: ChatGPT
- 🧪 Data Provider: ChatGPT
- 💻 Code Contributor: ChatGPT